
Very interesting and may be so helpful.
Below is a direct quote from the article below. I wonder why we are not told to add vitamin and mineral as well as OTC anti-inflammatory supplementation to out diets and medicine regimen? It would be worth it to try it, if it doesn’t help, ok, but imagine it does help! I am going to ask my psychiatrist about all of these and if they have any adverse reactions with the medications I’m on. That’s would be the best way to go here. Wishing us all the best of physical and mental health.
“It is now known that many mental health conditions are caused by inflammation in the brain which ultimately causes our brain cells to die. This inflammatory response starts in our gut and is associated with a lack of nutrients from our food such as magnesium, omega-3 fatty acids, probiotics, vitamins and minerals that are all essential for the optimum functioning of our bodies. Recent research has shown that food supplements such as zinc, magnesium, omega 3, and vitamins B and D3 can help improve people’s mood, relieve anxiety and depression and improve the mental capacity of people with Alzheimer’s. Magnesium is one of most important minerals for optimal health, yet many people are lacking in it. One study found that a daily magnesium citrate supplement led to a significant improvement in depression and anxiety, regardless of age, gender or severity of depression. Improvement did not continue when the supplement was stopped. Omega-3 fatty acids are another nutrient that is critical for the development and function of the central nervous system – and a lack has been associated with low mood, cognitive decline and poor comprehension. Research has shown that supplements like zinc, magnesium and vitamins B and D can improve the mental capacity of people with Alzheimer’s. Shutterstock The role of probiotics – the beneficial live bacteria in your digestive system – in improving mental health has also been explored by psychiatrists and nutritionists, who found that taking them daily was associated with a significant reduction in depression and anxiety. Vitamin B complex and zinc are other supplements found to reduce the symptoms of anxiety and depression. Hope for the future? These over-the-counter” supplements are widely available in supermarkets, chemists and online health food stores, although the cost and quality may vary. For people who have not responded to prescription drugs or who cannot tolerate the side effects, nutritional intervention can offer hope for the future. There is currently much debate over the effectiveness of antidepressants. The use of food supplements offer an alternative approach that has the potential to make a significant difference to the mental health of all age groups.”
http://theconversation.com/why-nutritional-psychiatry-is-the-future-of-mental-health-treatment-92545
A lack of essential nutrients is known to contribute to the onset of poor mental health in people suffering from anxiety and depression, bipolar disorder, schizophrenia and ADHD. Nutritional psychiatry is a growing discipline that focuses on the use of food and supplements to provide these essential nutrients as part of an integrated or alternative treatment for mental health disorders. But nutritional approaches for these debilitating conditions are not widely accepted by mainstream medicine. Treatment options tend to be limited to official National Institute for Care Excellence (NICE) guidelines which recommend talking therapies and antidepressants. Use of antidepressants Antidepressant use has more than doubled in recent years. In England 64.7m prescriptions were issued for antidepressants in 2016 at a cost of £266.6m. This is an increase of 3.7m on the number of items prescribed in 2015 and more than double than the 31m issued in 2006. A recent Oxford University study found that antidepressants were more effective in treating depression than placebo. The study was led by Dr Andrea Cipriani who claimed that depression is under treated. Cipriani maintains that antidepressants are effective and a further 1m prescriptions should be issued to people in the UK. This approach suggests that poor mental health caused by social conditions is viewed as easily treated by simply dispensing drugs. But antidepressants are shunned by people whom they could help because of the social stigma associated with mental ill-health which leads to discrimination and exclusion. Prescriptions for 64.7m items of antidepressants were dispensed in England in 2016, the highest level recorded by the NHS. Shutterstock More worrying is the increase in the use of antidepressants by children and young people. In Scotland, 5,572 children under 18 were prescribed antidepressants for anxiety and depression in 2016. This figure has more than doubled since 2009/2010. But according to British psychopharmacologist Professor David Healy, 29 clinical trials of antidepressant use in young people found no benefits at all. These trials revealed that instead of relieving symptoms of anxiety and depression, antidepressants caused children and young people to feel suicidal. Healy also challenges their safety and effectiveness in adults. He believes that antidepressants are over-prescribed and that there is little evidence that they are safe for long-term use. Antidepressants are said to create dependency, have unpleasant side effects and cannot be relied upon to always relieve symptoms. Nutrition and poor mental health In developed countries such as the UK we eat a greater variety of foodstuffs than ever before – but it doesn’t follow that we are well nourished. In fact, many people do not eat enough nutrients that are essential for good brain health, opting for a diet of heavily processed food containing artificial additives and sugar. The link between poor mental health and nutritional deficiencies has long been recognised by nutritionists working in the complementary health sector. However, psychiatrists are only now becoming increasingly aware of the benefits of using nutritional approaches to mental health, calling for their peers to support and research this new field of treatment. It is now known that many mental health conditions are caused by inflammation in the brain which ultimately causes our brain cells to die. This inflammatory response starts in our gut and is associated with a lack of nutrients from our food such as magnesium, omega-3 fatty acids, probiotics, vitamins and minerals that are all essential for the optimum functioning of our bodies. Recent research has shown that food supplements such as zinc, magnesium, omega 3, and vitamins B and D3 can help improve people’s mood, relieve anxiety and depression and improve the mental capacity of people with Alzheimer’s. Magnesium is one of most important minerals for optimal health, yet many people are lacking in it. One study found that a daily magnesium citrate supplement led to a significant improvement in depression and anxiety, regardless of age, gender or severity of depression. Improvement did not continue when the supplement was stopped. Omega-3 fatty acids are another nutrient that is critical for the development and function of the central nervous system – and a lack has been associated with low mood, cognitive decline and poor comprehension. Research has shown that supplements like zinc, magnesium and vitamins B and D can improve the mental capacity of people with Alzheimer’s. Shutterstock The role of probiotics – the beneficial live bacteria in your digestive system – in improving mental health has also been explored by psychiatrists and nutritionists, who found that taking them daily was associated with a significant reduction in depression and anxiety. Vitamin B complex and zinc are other supplements found to reduce the symptoms of anxiety and depression. Hope for the future? These over-the-counter” supplements are widely available in supermarkets, chemists and online health food stores, although the cost and quality may vary. For people who have not responded to prescription drugs or who cannot tolerate the side effects, nutritional intervention can offer hope for the future. There is currently much debate over the effectiveness of antidepressants. The use of food supplements offer an alternative approach that has the potential to make a significant difference to the mental health of all age groups. The emerging scientific evidence suggests that there should be a bigger role for nutritional psychiatry in mental health within conventional health services. If the burden of mental ill health is to be reduced, GPs and psychiatrists need to be aware of the connection between food, inflammation and mental illness. Medical education has traditionally excluded nutritional knowledge and its association with disease. This has led to a situation where very few doctors in the UK have a proper understanding of the importance of nutrition. Nutritional interventions are thought to have little evidence to support their use to prevent or maintain well-being and so are left to dietitians, rather than doctors, to advise on. But as the evidence mounts up, it is time for medical education to take nutrition seriously so that GPs and psychiatrists of the future know as much about its role in good health as they do about anatomy and physiology. The state of our mental health could depend on it.

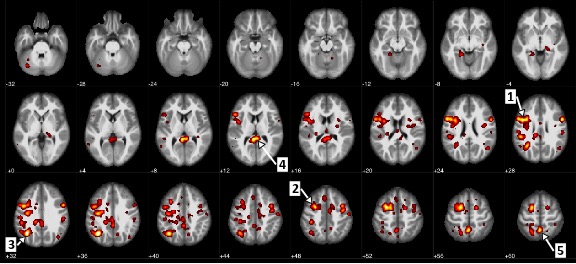


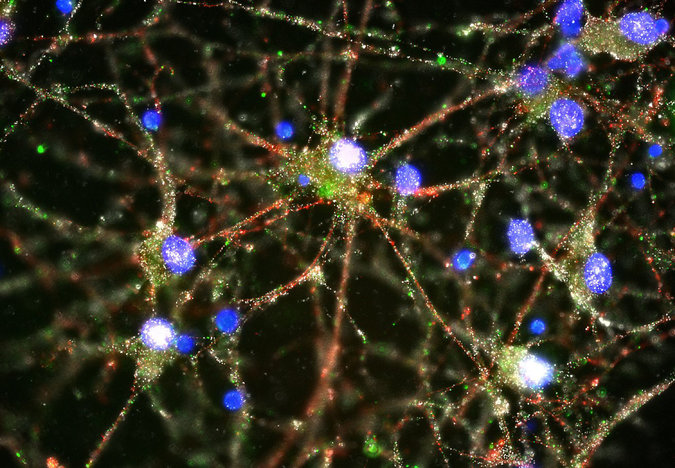 This is absolutely amazing! Scientists have found out that the cause of schizophrenia is too much synaptic pruning of neurons, “in which the brain sheds weak or redundant connections between neurons as it matures” in the prefrontal cortex. This begins in adolescence and the symptoms of schizophrenia also start in adolescence! “People who carry genes that accelerate or intensify that pruning are at higher risk of developing schizophrenia than those who do not, the new study suggests.”
This is absolutely amazing! Scientists have found out that the cause of schizophrenia is too much synaptic pruning of neurons, “in which the brain sheds weak or redundant connections between neurons as it matures” in the prefrontal cortex. This begins in adolescence and the symptoms of schizophrenia also start in adolescence! “People who carry genes that accelerate or intensify that pruning are at higher risk of developing schizophrenia than those who do not, the new study suggests.”



